EUDAMED Overview のページにある、EUDAMED TimeLine資料が、2025年11月27日に更新されました。今回の更新内容は、2025年11月26日委員会採決、11月27日公布のOJEU(Commission Decision (EU) 2025/2371)に基づき修正された内容と考えられます。
2024年6月13日に公布された「Regulation (EU) 2024/1860」 の第1条第6項(Article 1 (6) Article 123(3) is amended) のMDR 第123条第3項(IVDR 第113条3項)の改訂として、
MDR原文(英語版):
the introductory wording is replaced by the following:
‘without prejudice to the obligations of the Commission pursuant to Article 34, the obligations and requirements that relate to any of the electronic systems referred to in Article 33(2) shall apply from the date corresponding to 6 months from the date of publication of the notice referred to in Article 34(3), informing that the relevant electronic system is functional and meets the functional specifications drawn up pursuant to Article 34(1). The provisions referred to in the preceding sentence are:’
MDR日本語訳:
「委員会の第34条に基づく義務に影響を及ぼすことなく、第33条(2)に規定する電子システムに関する義務及び要件は、当該電子システムが機能しており、かつ、第34条(1)に基づいて作成された機能仕様を満たしていることを通知する第34条(3)に規定する通知の公表の日から6ヶ月後の日から適用される。前文に規定する内容は、次のとおりである。」
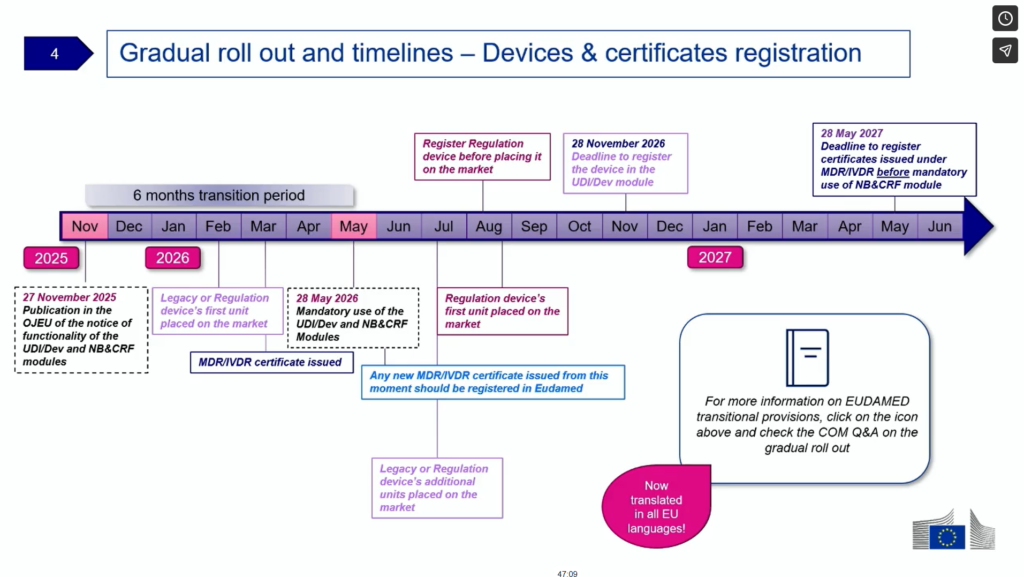
という箇所から、公示日(Date of Publication(DoP)=2025年11月27日(木))の6か月後の2026年5月28日(木)(Date of Mandatory(DoM))から、MDRの法的要件としてEUDAMEDへのUDIデバイス情報登録が義務化されます。ただし、公示日(2025年11月26日)から12か月以内の登録条件として、以下の条文が加えられているため、2026年5月28日以降もEU市場で継続されているデバイスは、EUDAMEDへのUDI登録を、公布日から12か月以内(=2026年11月28日まで)に完了することが、法的要件となります。
MDR原文(英語版):
(b)point (e) is replaced by the following:
‘(e)no later than 12 months from the date of publication of the notice referred to in Article 34(3) in respect of the electronic system referred to in Article 33(2), points (a) and (b), manufacturers shall ensure that the information to be entered in Eudamed in accordance with Article 29 is entered in that electronic system, including regarding the following devices, provided that those devices are also placed on the market from 6 months from the date of publication of that notice:
(i)devices, other than custom-made devices, for which the manufacturer has undertaken a conformity assessment in accordance with Article 52;
(ii)devices, other than custom-made devices, placed on the market pursuant to Article 120(3), (3a) or (3b), unless the device, for which the manufacturer has undertaken a conformity assessment in accordance with Article 52, is already registered in Eudamed;’
MDR日本語訳:
(b) (e) は次の文言に置き換えられる。
「(e) 製造業者は、第33条第2項(a)及び(b)に規定する電子システムに関する第34条第3項に規定する通知の公示の日から12ヶ月以内に、第29条に従ってEudamedに入力されるべき情報が、当該電子システムに入力されることを確保するものとする。これには、次に掲げる機器に関する情報も含まれる。ただし、当該通知の公示の日から6ヶ月以降も当該機器が市場ある場合に限る。
(i) 製造業者が第52条の規定に従って適合性評価を実施したカスタムメイド機器以外の機器。
(ii) 第120条第3項(3a)又は(3b)の規定に従って市場に投入されるカスタムメイド機器以外の機器。ただし、製造業者が第52条の規定に従って適合性評価を実施した機器が既にEudamedに登録されている場合を除く。」
12月3日に開催されたEUDAMED WORKSHOPで説明された資料がこちらになります。DoM(2026年5月28日)以前に上市(2026年2月中旬の箱)され、DoM以降に追加で出荷(2026年7月中旬の箱)される製品のDeadLine(=登録期限)は、2026年11月28日までとの記載があり、また、DoM以降が上市の場合(2026年8月下旬の箱)は、上市前(2026年8月上旬の箱)にEUDAMEDへの登録が必須と説明されています